Ayuda para leer cromatograma
paz
En este cromatograma se encuentra una variación genética:

Dice que la "secuencia de referencia" es la línea superior y que puedo usar el código genético general para encontrar el marco de lectura.
Puedo ver que hay dos N en lugar de una G y una C. Sin embargo, no sé qué significa esto. Tampoco estoy seguro de cómo encontraría el marco de lectura. Supongo que podría buscar un codón de inicio y finalización, pero ¿podría leerlos en ambos sentidos?
Última pregunta: ¿Es probable que este cambio sea patogénico? No estoy seguro de cómo clasificar una variación como patógena o no.
Respuestas (2)
cagliari2005
Los rastros que has obtenido proceden de la secuenciación de Sanger . N en genética significa nucleótido (¿sorprendente, verdad?). N se usa cuando se desconoce la base en una ubicación dada (o podría ser cualquier par de bases).
En su caso, tiene N porque el software de llamada de base no puede determinar el nucleótido. La primera N se debe a la superposición de dos picos (señales G y A) y la segunda debería ser una C en lugar de una N.
Por lo que puedo ver, no tiene mutaciones aparentes, por lo que es poco probable que esos cambios sean patógenos. Se puede llamar a una mutación cuando sabe que una base en la referencia se modifica en su muestra, lo que no es el caso aquí (simplemente N -> G y N -> C). Las señales de su N se ven muy similares entre su referencia y su muestra, por lo que no sugieren una mutación.
Para el marco de lectura, debe buscar un codón de inicio (ATG) y un codón de finalización (TAG, TAA o TGA) para identificar el marco de lectura abierto (ORF). Múltiples programas hacen eso y aquí hay un enlace a una herramienta NCBI en línea llamada ORF Finder .
Como señaló, el ORF también puede estar en el hilo inverso. No quieres mirar la secuencia inversa sino la secuencia complementaria inversa . Por lo general, las herramientas para detectar ORF tienen la opción de observar ambos hilos.
el hombre de la noche
Entonces, las dos N que ves no son necesariamente variantes, sino que probablemente solo sean lecturas de mala calidad. Esencialmente, cuando secuencias el ADN y el secuenciador no puede hacer una llamada sobre cuál es la base, simplemente la designará como N, lo que significa que la base podría ser cualquiera de las cuatro bases de ADN.
En cuanto a encontrar la lectura de, si sabe que esta secuencia contiene un codón de parada, eso ayuda, y simplemente busque cualquiera de las secuencias de parada. De lo contrario, busque bases que coincidan con las secuencias de codones y si obtiene una hebra completa de secuencia que codifica aminoácidos, es probable que tenga razón.
Como referencia , aquí hay un enlace a un gráfico de codones que lo ayudará a descifrar el marco de lectura.
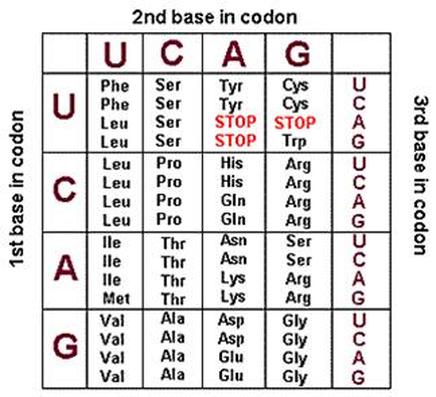{kind=link}
¿Eso ayuda?
Escribe los haplotipos de la familia.
Problema de pedigrí y tipo de herencia [cerrado]
El pedigrí del trastorno X recesivo me confunde
Problema de secuenciación de ADN
Teorema de Bayes para mutaciones
Trastornos X-recesivos y marcadores genéticos
¿Qué es un marcador genético?
Mutaciones de ADN en células de mamífero CHO-KI
Pregunta sobre los alelos autosómicos recesivos
¿Puede la prueba de ADN del hermano de mis abuelos revelar mi herencia de esa rama de la familia?
paz
el hombre de la noche